Amber: LEAP to prepare force field and initial coordinate input files
I. Concepts
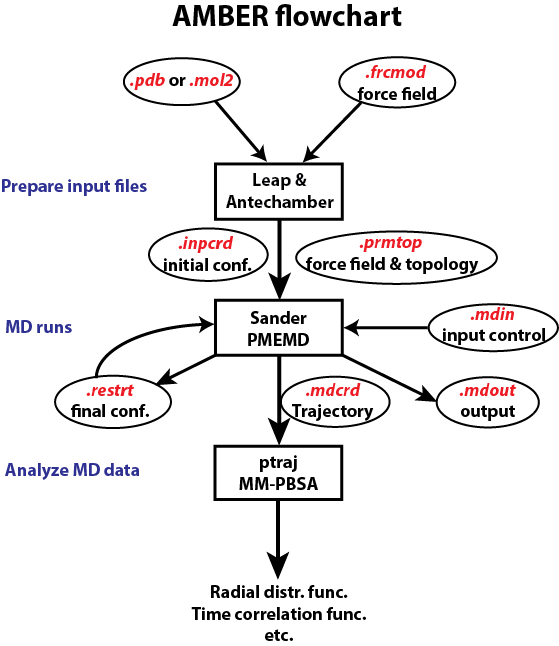
In this note, we discuss how to use LEAP to generate force field (prmtop) and initial coordinate (inpcrd) input files for Amber MD program sander. We will have to convert files to Amber-suited PDB/mol2 and frcmod files. (Note: Leap commands are not case sensitive, like loadamberparams and loadAmberParams are all OK; but object variables are case sensitive, like WAT, wat, Wat are not the same thing.)
TABLE 1. The information contained in different files.
| Information | PDB | mol2 | lib/prep | frcmod/dat | prmtop | inpcrd |
|---|---|---|---|---|---|---|
| Residue/Unit Name | Y | Y | Y | Y | ||
| Atom Name | Y | Y | Y | Y | ||
| Atom Type | Y | Y | Y | Y | ||
| Charges | Y | Y | Y | |||
| Connectivity | D | Y | Y | Y | ||
| Coordinates | Y | Y | D | Y | ||
| Atom Mass | Y | Y | ||||
| Bonded param | Y | Y | ||||
| Non-bonded param | Y | Y |
Here, Y means yes, and D means will be discarded when converted to force field files.
The general idea of converting files are outlines below.
Standard residue (PDB + lib -> prmtop + inpcrd)
For standard residue in protein and nucleic acids PDB files, LEAP has lib standard force field files, so leap will merge information in PDB and lib by matching residue name and atom name. Also, leap will merge information in lib and frcmod/dat by matching atom type. Then leap generates topology (prmtop) and coordinate (inpcrd) files.
Non-standard ligands or molecules (PDB/mol2 + frcmod -> prmtop + inpcrd)
For non-standard ligands or molecules, we need to create force field library files by ourselves.
First, we use antechamber to convert PDB into mol2/prep files (note: connectivity in PDB is discarded. Coordinates in prep will be discarded).
Second, we use parmchk to construct new additional frcmod file for the non-standard molecules with the input of mol2/prep.
Third, leap will merge information in PDB and mol2/prep by matching residue name and atom name. Also, leap will merge information in mol2/prep, standard frcmod/dat, and the new additional frcmod by matching atom type. Finally, leap will generate prmtop and inpcrd files.
II. General procedure
(1) Single molecular structure
Draw molecule in ChemDraw or iQmol and save as .mol2 or .pdb format (Spartan can open .mol created by ChemDraw and save as .mol2 or .pdb files)
(2) Charge methods
| charge method | abbre. | index | charge method | abbre. | index |
|---|---|---|---|---|---|
| RESP | resp | 1 | AM1-BCC | bcc | 2 |
| CM1 | cm1 | 3 | CM2 | cm2 | 4 |
| ESP (Kollman) | esp | 5 | Mulliken | mul | 6 |
| Gasteiger | gas | 7 | Read in charge | rc | 8 |
| Write out charge | wc | 9 | Delete Charge | dc | 10 |
Option 1: Gaussian charge method like RESP
$ antechamber -i xx.ini.mol2 -fi mol2 -o xx.mol2 -fo mol2 -rn xx (refresh mol2 format for antechamber)
$ antechamber -i xx.mol2 -fi mol2 -o xx.gcrt -fo gcrt (generate g09 input file)
$ g09 < xx.gcrt > xx.gout (Gaussian calculation of charges)
$ antechamber -i xx.gout -fi gout -o xx.mol2 -fo mol2 -c resp -rn xx (assign charge resp)
(note that .mol2 file can be changed manually to match literature model values)
Option 2: semi-empirical method like bcc (AM1-BCC method)
$ antechamber -i xx.ini.mol2 -fi mol2 -o xx.mol2 -fo mol2 -c bcc -rn xx
(3) Use parmchk to convert mol2 (only) into frcmod
$ parmchk -i xx.mol2 -f mol2 -o xx.frcmod -a Y
$ vmd xx.mol2 (view graphics of molecule)
(4) Use LEAP to prepare inpcrd and prmtop files
A. Load molecules: xx.frcmod (the force field file) and xx.mol2 (the molecular geometry file)
copy both files to your leap folder, then $ xleap
> source leaprc.gaff (the general Amber force field, no topology)
other force fields options:
source leaprc.ff02pol
/export/apps/Amber/12/dat/leap/parm/frcmod.ff02pol.r1 (polarizable force field) ff03.r1
source leaprc.ff03.r1
/export/apps/Amber/12/dat/leap/parm/frcmod.ff03 (the non-polarizable force field)
Then create molecule called xx, yy,… and load force field parameters for xx,yy,…
> xx = loadmol2 xx.mol2
> loadamberparams xx.frcmod
> yy = loadmol2 yy.mol2
> loadamberparams yy.frcmod
> list
B. Solvate your molecule with solvents and form a simulation box
Option 1:
> loadoff solvents.lib (the default solvent models)
> list
CHCL3BOX DC4 MEOHBOX NMABOX PL3 POL3BOX QSPCFWBOX SPC
SPCBOX SPCFWBOX SPF SPG T4E TIP3PBOX TIP3PFBOX TIP4PBOX
TIP4PEWBOX TIP5PBOX TP3 TP4 TP5 TPF
Option 2:
Use solvateBox
> solvateBox xx yy 15
> saveAmberParms xx xx.prmtop xx.inpcrd
(here, xx is the solute and xx.prmtop and xx.inpcrd can be renamed afterwards)
> quit
Two input files for sander .prmtop and .inpcrd .prmtop is the force field file including both parameters and topology information for the whole solute/solvent system
.inpcrd is the initial configuration file
Option 3: use a script that loads all the input files except for the solvation step
e.g. $ xleap
> source script
the script file is as follows:
source leaprc.ff03.r1 OR source leaprc.ff02pol.r1
loadoff solvents.lib
hod = loadmol2 hod.mol2
leadAmberParams hod.frcmod
WAT = SPC
loadAmberParams frcmod.spce
list
then you type to finish the solvation step in xleap:
> solvateBox hod SPC 12.5
OR to use the default solvent box:
> solvateBox SPC MEOHBOX 12.0
> addIons CYN K+ 9
> saveAmberParams SPC (the solute) spc.prmtop spc.inpcrd
> quit
$ vmd New Molecule … –> load xx.prmtop using format AMBER7 Parm , then load xx.inpcrd using formate AMBER7 Restart to view the 3D structure of the system.
III. Example: Preparation of the PCBM_DMA in Chrorobenzene solution
Take the example of PCBM_DMA (residue name: PCB) as solute and Chlorobenzene (residue name: CBZ) as solvent. Three letter acronyms for residue name is preferred. We have PCBM_DMA.pdb CBZ.pdb data files.
(1) Convert pdb to mol2 using antechamber
For example, we use AM1-BCC empirical charge method, such that the missing charge information will be added to pdb file and form mol2 file.
$ antechamber -i CBZ.pdb -fi pdb -o CBZ.mol2 -fo mol2 -c bcc -s 2
In CBZ_SINGLE.pdb, “ATOM” is recode type, followed by atom serial number, name, residue name, x, y, z, occupancy, temperature factor. It is important to note “TER” marks the end of the residue that is required in Amber. “END” marks the end of the file. “ATOM” and “HEATOM” (for non-standard groups) are both ok.
ATOM 1 H CBZ 1 0.364 -2.147 0.000 1.00 0.00
ATOM 2 C CBZ 1 -0.185 -1.212 0.000 1.00 0.00
ATOM 3 C1 CBZ 1 -1.577 -1.204 0.000 1.00 0.00
ATOM 4 H1 CBZ 1 -2.111 -2.149 0.000 1.00 0.00
ATOM 5 C2 CBZ 1 -2.275 0.000 0.000 1.00 0.00
ATOM 6 H2 CBZ 1 -3.361 0.000 0.000 1.00 0.00
ATOM 7 C3 CBZ 1 -1.577 1.204 0.000 1.00 0.00
ATOM 8 H3 CBZ 1 -2.111 2.149 0.000 1.00 0.00
ATOM 9 C4 CBZ 1 -0.185 1.212 0.000 1.00 0.00
ATOM 10 H4 CBZ 1 0.364 2.147 0.000 1.00 0.00
ATOM 11 C5 CBZ 1 0.495 0.000 0.000 1.00 0.00
ATOM 12 Cl CBZ 1 2.247 0.000 0.000 1.00 0.00
TER
END
PDB file record format:
COLUMNS DATATYPE FIELD DEFINITION
-------------------------------------------------------------------------------------
1 - 6 Record name "ATOM "
7 - 11 Integer serial Atom serial number. (index=serial-1)
13 - 16 Atom name Atom name. (usually start from column 14!)
17 Character altLoc Alternate location indicator.
18 - 20 Residue name resName Residue name.
22 Character chainID Chain identifier.
23 - 26 Integer resSeq Residue sequence number.
27 AChar iCode Code for insertion of residues.
31 - 38 Real(8.3) x Orthogonal coordinates for X in Angstroms.
39 - 46 Real(8.3) y Orthogonal coordinates for Y in Angstroms.
47 - 54 Real(8.3) z Orthogonal coordinates for Z in Angstroms.
55 - 60 Real(6.2) occupancy Occupancy.
61 - 66 Real(6.2) tempFactor Temperature factor.
77 - 78 LString(2) element Element symbol, right-justified.
79 - 80 LString(2) charge Charge on the atom. (usually discarded)
The resulting CBZ.mol2 contains the 3 dimensional structure of our CBZ molecule as well as the charge on each atom, final column, the atom number (column 1), its name (column 2) and it’s atom type (column 6). It also specifies the bonding at the end of the file. This file does not, however, contain any parameters. The GAFF parameters are all defined in $AMBERHOME/dat/leap/parm/gaff.dat. The other thing you should notice here is that all of the GAFF atom types are in lower case. This is the mechanism by which the GAFF force field is kept independent of the macromolecular AMBER force fields. All of the traditional AMBER force fields use uppercase atom types. In this way the GAFF and traditional force fields can be mixed in the same calculation.
@<TRIPOS>MOLECULE
CBZ
12 12 1 0 0
SMALL
bcc
@<TRIPOS>ATOM
1 H 0.3640 -2.1470 0.0000 ha 1 CBZ 0.192810
2 C -0.1850 -1.2120 0.0000 ca 1 CBZ -0.158699
3 C1 -1.5770 -1.2040 0.0000 ca 1 CBZ -0.164546
4 H1 -2.1110 -2.1490 0.0000 ha 1 CBZ 0.178559
5 C2 -2.2750 0.0000 0.0000 ca 1 CBZ -0.163617
6 H2 -3.3610 0.0000 0.0000 ha 1 CBZ 0.174616
7 C3 -1.5770 1.2040 0.0000 ca 1 CBZ -0.164546
8 H3 -2.1110 2.1490 0.0000 ha 1 CBZ 0.178559
9 C4 -0.1850 1.2120 0.0000 ca 1 CBZ -0.158699
10 H4 0.3640 2.1470 0.0000 ha 1 CBZ 0.192810
11 C5 0.4950 0.0000 0.0000 ca 1 CBZ -0.090750
12 Cl 2.2470 0.0000 0.0000 cl 1 CBZ -0.016500
@<TRIPOS>BOND
1 1 2 1
2 2 3 ar
3 2 11 ar
4 3 4 1
5 3 5 ar
6 5 6 1
7 5 7 ar
8 7 8 1
9 7 9 ar
10 9 10 1
11 9 11 ar
12 11 12 1
@<TRIPOS>SUBSTRUCTURE
1 CBZ 1 TEMP 0 **** **** 0 ROOT
Do the same to the solute, and obtain PCBM_DMA.mol2 file.
(2) Convert mol2 to frcmod using parmchk
$ parmchk -i CBZ.mol2 -f mol2 -o CBZ.frcmod
The resulting CBZ.frcmod (force field modification type) file contains all the missing fore field parameters. If antechamber can’t empirically calculate a value or has no analogy it will either add a default value that it thinks is reasonable or alternatively insert a place holder (with zeros everywhere) and the comment “ATTN: needs revision”. In this case you will have to manually change parameters in this frcmod file. The CBZ.frcmod shows that there were 1 missing improper dihedral parameters.
remark goes here
MASS
BOND
ANGLE
DIHE
IMPROPER
ca-ca-ca-ha 1.1 180.0 2.0 General improper torsional angle (2 general atom types)
NONBON
Do the same to the solute, and obtain PCBM_DMA.frcmod file.
(3) Packmol : the program to create the simulation box by solvating the solute
$ packmol < setup.packm.PCBM
The packmol script setup.packm.PCBM is as follows.
# A mixture of PCBM and CBZ
# All the atoms from different molecules will be separated at least 2.0 Angstroms
tolerance 2.0
# The file type of input and output files is PDB
filetype pdb
# The name of the output file
output PCBM_CBZ_BULK.pdb
# 1 PCBM molecule and 6000 CBZ molecules will be put in a box
# defined by the minimum coordinates x, y and z = 0. 0. 0. and maximum
# coordinates 90. 90. 90. That is, they will be put in a cube of side
# 40. (the keyword "inside cube 0. 0. 0. 90.") could be used as well.
structure PCBM_DMA.pdb
number 1
inside box 35. 35. 35. 55. 55. 55.
end structure
structure CBZ.pdb
number 6000
inside box 0. 0. 0. 90. 90. 90.
end structure
(i) Use VMD to check visualize the box. In representation, Resname PCB and Resname CBZ to change rendering. OR use Jmol.sh to see structure.
(ii) Make sure net charge = 0.000000000 using dipole_read.f
(iii) Use UCSF Chimera to compare quantum .cube surface with the partial charges.
(4) LEAP to prmtop and inpcrd
A. Use leap to correct the BULK system PDB file into Amber acceptable PDB format.
One of the difference is the new PDB file has “TER” after every molecule/residue.
$ tleap -s -f setup.amber.INIT
setup.amber.INIT
source leaprc.gaff
CBZ = loadmol2 CBZ.mol2
loadamberparams CBZ.frcmod
PCB = loadmol2 PCBM_DMA.mol2
loadamberparams PCBM_DMA.frcmod
BULK = loadpdb PCBM_CBZ_BULK.pdb
savepdb BULK PCBM_CBZ_BULK_NEW.pdb
quit
B. Use leap to construct prmtop and inpcrd from the input of the NEW PDB of the whole BULK system, the mol2 of each species, and the additional force field parameter frcmod files.
We run the following leap script via tleap or via graphic interface xleap:
$ tleap -s -f setup.amber.BULK
setup.amber.BULK
source leaprc.gaff
CBZ = loadmol2 CBZ.mol2
loadamberparams CBZ.frcmod
PCB = loadmol2 PCBM_DMA.mol2
loadamberparams PCBM_DMA.frcmod
BULK = loadpdb PCBM_CBZ_BULK_NEW.pdb
setbox BULK centers
saveAmberParm BULK PCBM_CBZ_6001.prmtop PCBM_CBZ_6001.inpcrd
quit
Leave a Comment